Related Subjects:
|Dementias
|Gerstmann-Straussler-Scheinker Syndrome (GSS)
|Fatal Familial Insomnia (FFI)
|Creutzfeldt Jakob disease (CJD)
|Variant Creutzfeldt Jakob disease (vCJD)
|Kuru
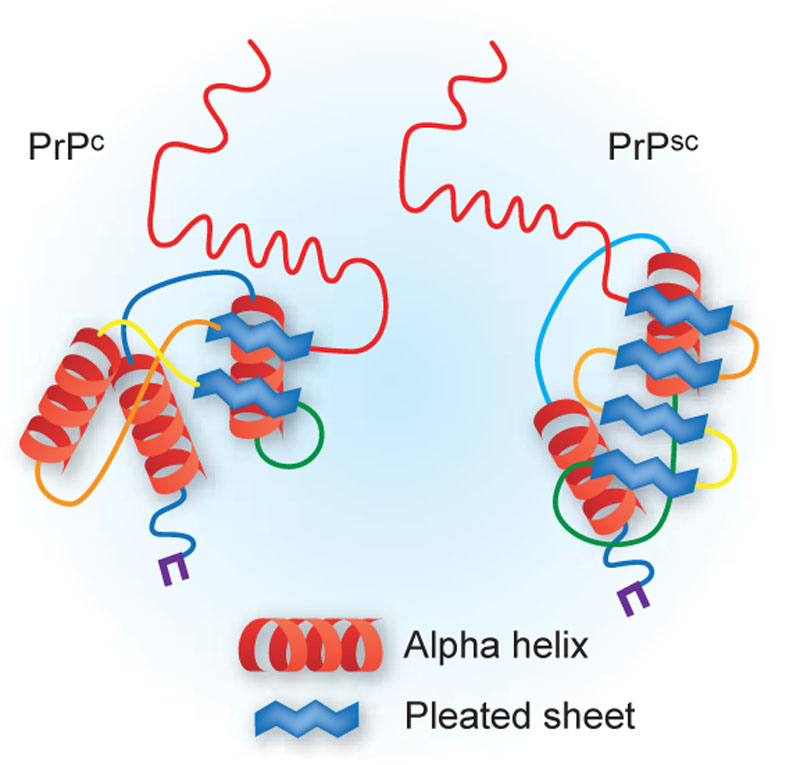
Pathophysiologically, CJD is a prion disease: misfolded prion protein triggers abnormal folding of native prion protein, causing rapidly progressive neurodegeneration, spongiform change, neuronal loss and gliosis. Clinically think of rapidly progressive dementia, psychiatric or behavioural change, ataxia, myoclonus, pyramidal/extrapyramidal signs, and akinetic mutism late on.
🧾 About
- The classical form of Creutzfeldt-Jakob Disease (CJD) is a prion disease unrelated to bovine spongiform encephalopathy (BSE).
- It affects mainly older adults and follows a very aggressive course compared with the variant form.
- Caused by misfolded prion proteins (PrPSc) that induce abnormal folding of normal host proteins (PrPC), leading to neurodegeneration.
🔎 Types
- Sporadic CJD (sCJD): Most common form (~85-90%).
- Familial CJD: 5–15%, autosomal dominant inheritance (mutations in PRNP gene).
- Iatrogenic CJD: Transmission through contaminated medical procedures (pituitary extracts, corneal transplants, dura mater grafts).
- Variant CJD (vCJD): Linked to BSE (“mad cow disease”), affecting younger patients.
📋 Diagnostic Criteria
- Definite: Neuropathology, immunocytochemistry, or Western blot confirming prion protein (PrPSc).
- Probable:
- Positive RT-QuIC (real-time quaking-induced conversion) in CSF or tissue OR
- Rapidly progressive dementia + ≥2 of:
- Myoclonus
- Visual/cerebellar disturbance
- Pyramidal or extrapyramidal features
- Akinetic mutism
- AND one of:
- EEG with periodic sharp-wave complexes
- CSF 14-3-3 protein (within 2 years of onset)
- MRI: High signal in caudate/putamen or cortical regions on DWI/FLAIR
- Possible: Dementia + ≥2 clinical features listed above, without confirmatory investigations.
🧠 Diagram
⚖️ Classic vs Variant CJD
| Characteristic | Classic CJD | Variant CJD |
|---|
| Median age at death | 68 years | 28 years |
| Duration of illness | 4–5 months | 13–14 months |
| Clinical onset | Dementia; early neurologic signs | Psychiatric/behavioral symptoms, painful dysesthesias |
| EEG | Periodic sharp waves often present | Often absent |
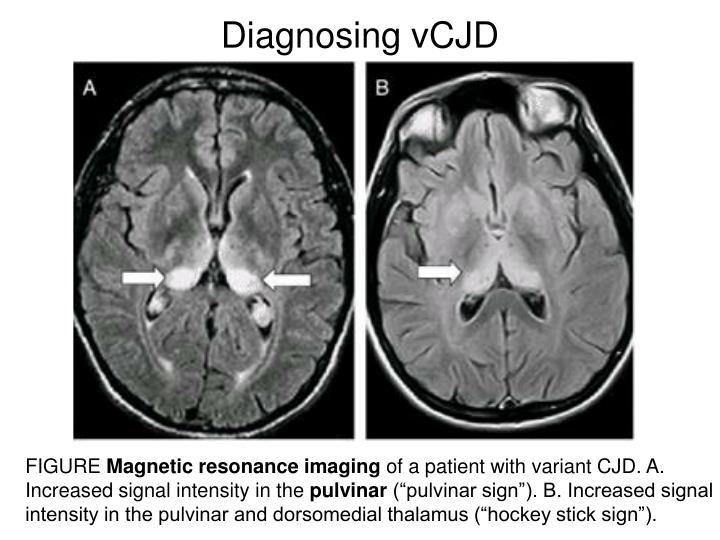
| MRI | Basal ganglia hyperintensity | Pulvinar sign (>75%) |
| Neuropathology | Spongiform change ± rare plaques | Florid plaques common |
| Lymphoid tissue prions | Not detected | Readily detected |
🩺 Clinical Presentation
- Rapidly progressive dementia with myoclonus (classic triad).
- Other features: ataxia, visual disturbance, behavioral change, extrapyramidal signs.
- Variant CJD: Younger patients, psychiatric prodrome, delayed dementia.
🧪 Investigations
- EEG: Periodic triphasic sharp-wave complexes (seen in ~2/3 of sCJD).
- MRI: DWI/FLAIR hyperintensity in caudate, putamen, or cortical ribboning. 🏒 Hockey stick sign = bilateral pulvinar + dorsomedial thalamic MRI hyperintensity, classically seen in variant CJD.
- CSF: 14-3-3 protein (non-specific, can be positive in stroke/MS); RT-QuIC is more specific.
- Brain biopsy/post-mortem: Gold standard – spongiform change, neuronal loss, and PrPSc deposition.
💊 Management
- No curative treatment – management is supportive.
- Report suspected cases to the CJD surveillance unit (UK requirement).
- Genetic counseling for familial cases.
- Emphasis on infection control (prion proteins resist sterilisation).
