
| Download the amazing global Makindo app: ✅ Means NICE/National Guidelines 2026 compliant Android | Apple | |
|---|---|
| MEDICAL DISCLAIMER: Educational use only. Not for diagnosis or management. See below for full disclaimer. |
Crouzon Syndrome
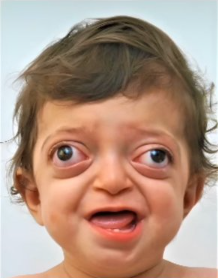
Crouzon syndrome is a syndromic craniosynostosis caused by premature fusion of one or more cranial sutures, leading to abnormal skull and facial development. It is classically associated with midface hypoplasia and shallow orbits (causing proptosis), but no limb anomalies (a key discriminator from Apert syndrome). In UK practice, suspected cases should be referred early to a specialist craniofacial MDT to assess for raised intracranial pressure, visual risk, airway compromise, hearing problems, and to plan staged surgical care.
✅ Core features
- Craniosynostosis: variable head shapes depending on sutures involved (often brachycephaly)
- Midface hypoplasia: maxillary underdevelopment → class III malocclusion, dental crowding
- Ocular features: shallow orbits → proptosis, exposure risk, strabismus
- Nasal shape: beaked nose, relative mandibular prognathism due to midface retrusion
- ENT: recurrent otitis media and conductive hearing loss can occur
- Neurological risk: raised intracranial pressure (ICP) in some children
- Limbs: no syndactyly (helps distinguish from Apert)
🧬 Genetics and pathophysiology
Crouzon syndrome is most commonly due to autosomal dominant mutations in FGFR2. FGFR2 signalling influences osteoblast differentiation and suture biology; activating mutations drive premature osteogenesis at cranial sutures, causing early fusion. The brain continues to grow, so skull expansion is forced into remaining patent sutures, producing characteristic head shapes and potentially reducing intracranial reserve, which explains the risk of raised ICP and secondary visual compromise.
🔎 Clinical assessment
- History: feeding/breathing difficulties, snoring/apnoea symptoms, headaches, irritability, developmental concerns
- Examination: head shape, midface retrusion, ocular proptosis/strabismus, dental occlusion, airway signs
- Red flags suggesting raised ICP: persistent headaches, vomiting, behavioural change, papilloedema, visual symptoms
- Hearing: screen for recurrent otitis media and hearing impairment
🖼️ Investigations
- CT skull with 3D reconstruction: confirms fused sutures and defines craniofacial anatomy for planning
- Ophthalmology assessment: vision testing, fundoscopy for papilloedema, corneal exposure risk
- Sleep/airway assessment: consider sleep study if obstructive symptoms
- Audiology: formal hearing testing
- Genetics: FGFR2 testing to confirm diagnosis and enable family counselling
🧠 Why raised ICP can happen
When sutures fuse early, skull growth is restricted in the direction perpendicular to the fused suture. The growing brain then drives compensatory skull expansion elsewhere, but in some children the mismatch between brain growth and cranial vault capacity contributes to raised ICP. Clinically, this matters because sustained raised ICP can damage the optic nerves, impair development, and worsen headaches; timely MDT surveillance and surgery aim to prevent these downstream consequences.
🛠️ Management (MDT)
- Craniofacial MDT referral: early specialist assessment (neurosurgery, plastics, ENT, ophthalmology, orthodontics, genetics)
- Surgery in infancy: cranial vault expansion/remodelling in selected cases to improve shape and reduce ICP risk
- Midface advancement (later childhood/adolescence): improves airway, ocular protection, and occlusion (e.g., Le Fort III)
- Ophthalmology: manage strabismus, monitor optic nerves, treat exposure keratopathy
- ENT/audiology: treat otitis media, consider grommets if indicated, address hearing loss
- Dental/orthodontics: manage malocclusion and crowding; plan alongside craniofacial surgery timeline
- Neurodevelopment: developmental surveillance and early intervention support if needed
🇬🇧 UK context
In the NHS, suspected craniosynostosis syndromes should be referred promptly to a regional specialist craniofacial centre for coordinated imaging, genetics, and staged surgical planning. Early detection is important because vision, airway, hearing, and neurodevelopmental outcomes depend on timely MDT surveillance and intervention. Families should be offered genetic counselling given autosomal dominant inheritance and the possibility of de novo variants.
🧩 Differentials (quick discriminator)
- Apert syndrome: craniosynostosis + syndactyly
- Pfeiffer syndrome: craniosynostosis + broad thumbs/toes
- Saethre-Chotzen: often milder craniofacial changes, low hairline, ptosis, limb changes can occur
📌 Exam and ward pearls
- Think vision and ICP in any craniosynostosis: ask about headaches and check fundoscopy where possible
- No limb anomalies strongly supports Crouzon over Apert
- Midface hypoplasia can be an airway problem: ask about snoring and apnoeas
| The content on this website is provided for educational and informational purposes only to support exam preparation (e.g., MLA, MRCP, USMLE) and learning. This is NOT medical advice, diagnosis, treatment, or professional guidance. It does not replace consultation with a qualified healthcare professional, official guidelines (e.g., NICE, GMC, BNF), or supervised clinical practice. Always verify information with current, authoritative sources. Makindo and its contributors accept no liability for any reliance on this content, including errors, omissions, or any resulting harm, loss, or consequences. By using this site, you agree to these terms. |
|
|
Categories
- About
- Acute Medicine
- Anaesthetics and Critical Care
- Anatomy
- Anatomy and Physiology
- Biochemistry
- Book
- Cardiology
- Collections
- CompSci
- Crib Sheets
- Critical care
- Dental
- Dermatology
- Differentials
- Drugs
- ENT
- Electrocardiogram
- Embryology
- Emergency Medicine
- Endocrinology
- Ethics
- Foundation Doctors
- GCSE
- Gastroenterology
- General Practice
- Genetics
- Geriatric Medicine
- Geriatrics
- Guidelines
- Haematology
- Hepatology
- Immunology
- Infectious Diseases
- Infographic
- Investigations
- Lists
- MRCP
- Mandatory Training
- Medical Students
- Microbiology
- Nephrology
- Neurology
- Neurosurgery
- Nutrition
- OSCE
- Obstetrics Gynaecology
- Oncology
- Ophthalmology
- Oral Medicine and Dentistry
- Orthopaedics
- Paediatrics
- Palliative
- Palliative Care
- Pathology
- Pharmacology
- Physiology
- Procedures
- Psychiatry
- Public Health
- Radiology
- Respiratory
- Resuscitation
- Revision Topics
- Rheumatology
- Statistics and Research
- Stroke
- Surgery
- Toxicology
- Trauma and Orthopaedics
- USMLE
- Urology
- Vascular Surgery