Myotonic dystrophy - Dystrophia myotonica
🧬 About
- Myotonic dystrophy (Dystrophia Myotonica) is the most common muscular dystrophy of adult life.
- It is an autosomal dominant condition with variable penetrance, showing progressive multisystem involvement.
⚖️ Aetiology
- Type 1 (DM1): Caused by expanded trinucleotide (CTG) repeat in the 3’ untranslated region of the DMPK gene (chromosome 19).
Abnormal if >34 repeats. Demonstrates anticipation → earlier, more severe onset in successive generations. Prenatal testing possible.
- Type 2 (DM2 / Proximal Myotonic Myopathy): Due to expanded CCTG repeat in an intronic region of the ZNF9 gene (chromosome 3).
Presents in young adults with proximal weakness (neck/finger flexors, hips), myotonia, cataracts. Generally milder than DM1.
- Anticipation: Disease severity increases down generations due to further repeat expansions.
🧑⚕️ Clinical Features
- Characteristic appearance: frontal balding, ptosis, facial muscle wasting (temporalis, SCM).
- Progressive muscle weakness and atrophy – usually mild and slowly progressive.
- Cardiac conduction defects (PR prolongation, arrhythmias) → may require pacemaker (distinct from cardiomyopathy seen in Duchenne/Becker).
- Cataracts (often early, “Christmas tree” opacities).
- Endocrine/metabolic: impaired glucose tolerance, insulin resistance.
- Respiratory muscle weakness → risk of sleep apnoea, hypoventilation.
- Cognitive involvement: learning difficulties, reduced IQ in ~50%.
- GI dysmotility: dysphagia, constipation, pseudo-obstruction.
- Immunological abnormalities: low IgG levels in some cases.
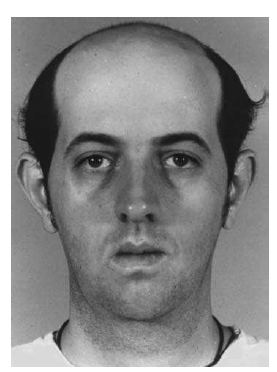
Young man with DM1: frontal baldness, bilateral ptosis, and wasting of temporalis, facial, and sternocleidomastoid muscles.
📊 Comparison of Myotonic Dystrophy Type 1 and Type 2
| Feature |
Type 1 (DM1) |
Type 2 (DM2) |
| Genetic Mutation |
CTG trinucleotide repeat in DMPK gene (chromosome 19) |
CCTG tetranucleotide repeat in ZNF9 gene (chromosome 3) |
| Onset |
Congenital, childhood, or adult-onset (variable) |
Typically young adulthood |
| Anticipation |
Prominent - worsens with each generation |
Mild/absent anticipation |
| Distribution of Weakness |
Distal & facial (ptosis, SCM, temporalis, hand muscles) |
Proximal (hip flexors, neck flexors, finger flexors) |
| Myotonia |
Prominent, often disabling |
Present but usually milder |
| Systemic Features |
Cataracts, insulin resistance, cardiac conduction defects, GI dysmotility, cognitive impairment |
Cataracts, muscle pain, less cardiac involvement, rare cognitive effects |
| Severity |
More severe, multisystem, congenital forms possible |
Milder, slower progression, rarely life-threatening |
🧪 Investigations
- Genetic Testing: Gold standard – identifies repeat expansion.
- CK (Creatine Kinase): May be mildly elevated (2–10× normal).
- Muscle Biopsy: Myofibre atrophy + central nuclei.
- EMG: Classic myotonic discharges (waxing and waning potentials).
- ECG: PR prolongation, atrial flutter, ventricular arrhythmias.
- Other labs: Low IgG; glucose tolerance tests.
💊 Management
- Myotonia: Phenytoin (100 mg TDS) preferred. Mexiletine sometimes used.
⚠️ No treatment halts progressive weakness.
- Cardiac: Annual ECG, 24-hr Holter, cardiology review. Pacemaker/ICD if significant conduction disease.
- Respiratory: Avoid respiratory depressants. Consider CPAP for sleep apnoea.
- Obstetric: Risk of ineffective uterine contractions; multidisciplinary delivery planning.
- Supportive: Speech/swallow therapy, physiotherapy, cataract surgery, endocrine screening.
🛑 Anaesthetic Precautions
- Avoid suxamethonium → prolonged contraction & dangerous myotonia.
- Non-depolarising relaxants = safe at usual doses; reversal agents tolerated.
- Mechanical/electrical stimuli (diathermy) may trigger myotonia → use caution.
- Regional anaesthesia often preferred.
📌 Key Points
- DM = a multisystem disorder (muscle, eye, heart, endocrine, respiratory, cognitive, GI).
- Anticipation leads to earlier/more severe disease in successive generations.
- Cardiac monitoring is essential → conduction disease is a major cause of sudden death.
- No curative therapy; management is supportive and multidisciplinary.
Cases - Myotonic Dystrophy (Dystrophia Myotonica)
- Case 1 - Adult with grip myotonia ✋: A 28-year-old man presents with difficulty releasing his grip after shaking hands. He also reports progressive weakness of facial and distal limb muscles. Exam: long thin face, frontal balding, ptosis, and “hatchet” facies. EMG: myotonic discharges. Diagnosis: Myotonic dystrophy type 1. Managed with symptomatic therapy (physio, speech therapy) and cardiac monitoring.
- Case 2 - Multisystem involvement ❤️👁️: A 40-year-old woman with known myotonic dystrophy presents with daytime somnolence, cataracts, and palpitations. ECG: conduction block. Exam: distal weakness, myotonia on percussion of thenar eminence. Diagnosis: multisystem complications of myotonic dystrophy. Managed with cataract surgery, pacemaker insertion if conduction disease, and endocrine screening (thyroid, diabetes).
- Case 3 - Congenital form 👶: A newborn boy is floppy at birth with severe hypotonia and respiratory distress. Mother has mild muscle weakness and early cataracts. Baby requires ventilatory support. Genetic testing: CTG repeat expansion in DMPK gene. Diagnosis: congenital myotonic dystrophy (maternal transmission). Managed with NICU care and long-term multidisciplinary follow-up.
Teaching Point 🩺: Myotonic dystrophy is the most common adult muscular dystrophy.
- Type 1 (DMPK gene, CTG repeat) → distal weakness, grip myotonia, cataracts, cardiac conduction defects.
- Type 2 (CNBP gene, CCTG repeat) → proximal weakness, later onset.
Inheritance: autosomal dominant, anticipation seen (earlier/severe disease in successive generations).
Always monitor for respiratory, cardiac, and endocrine complications.
