
| Download the amazing global Makindo app: ✅ Means NICE/National Guidelines 2026 compliant Android | Apple | |
|---|---|
| MEDICAL DISCLAIMER: Educational use only. Not for diagnosis or management. See below for full disclaimer. |
Myelofibrosis 🩸
Related Subjects: |Iron deficiency Anaemia |Haemolytic anaemia |Macrocytic anaemia |Megaloblastic anaemia |Microcytic anaemia |Myelodysplasia |Myelofibrosis |Hereditary Spherocytosis |Hereditary Elliptocytosis |Haemophilia A |Haemophilia B |Haemolytic anaemia |Heme |Globins |Red blood cells |White blood cells |Lymphocytes |Platelets |Cryoprecipitate |Fresh Frozen Plasma |Blood Cell Maturation |Blood film interpretation |Reticulocytes
🩸 Primary myelofibrosis is a myeloproliferative neoplasm where bone marrow is progressively replaced by fibrous tissue (reticulin and collagen). This leads to ineffective haematopoiesis, extramedullary haematopoiesis, and massive splenomegaly. ⚠️ A minority of cases transform to acute myeloid leukaemia (AML).
📖 About
- One of the BCR-ABL negative myeloproliferative neoplasms (alongside polycythaemia vera and essential thrombocythaemia).
- Characterised by marrow fibrosis, abnormal megakaryocyte proliferation, and extramedullary haematopoiesis.
- Median age of onset: 60–70 years.
- Can be primary or evolve secondarily from polycythaemia vera / essential thrombocythaemia.
🧬 Aetiology / Pathogenesis
- Clonal proliferation of megakaryocytes → release of platelet-derived growth factor (PDGF) and TGF-β → marrow fibrosis.
- Haematopoiesis shifts to spleen and liver → massive hepatosplenomegaly.
- Mutations: JAK2 V617F (~50%), CALR (~30%), MPL (~10%).
- Complications: anaemia, splenic infarction, portal hypertension, AML transformation (~10–20%).
🩺 Clinical Features
- Constitutional symptoms: fever, night sweats, weight loss (“B symptoms”).
- Massive splenomegaly (often palpable well below costal margin).
- Hepatomegaly (extramedullary haematopoiesis).
- Anaemia-related symptoms: fatigue, dyspnoea.
- Bone pain (marrow expansion / fibrosis).
- Complications: infections, gout (hyperuricaemia), thrombosis, bleeding.
🔍 Investigations
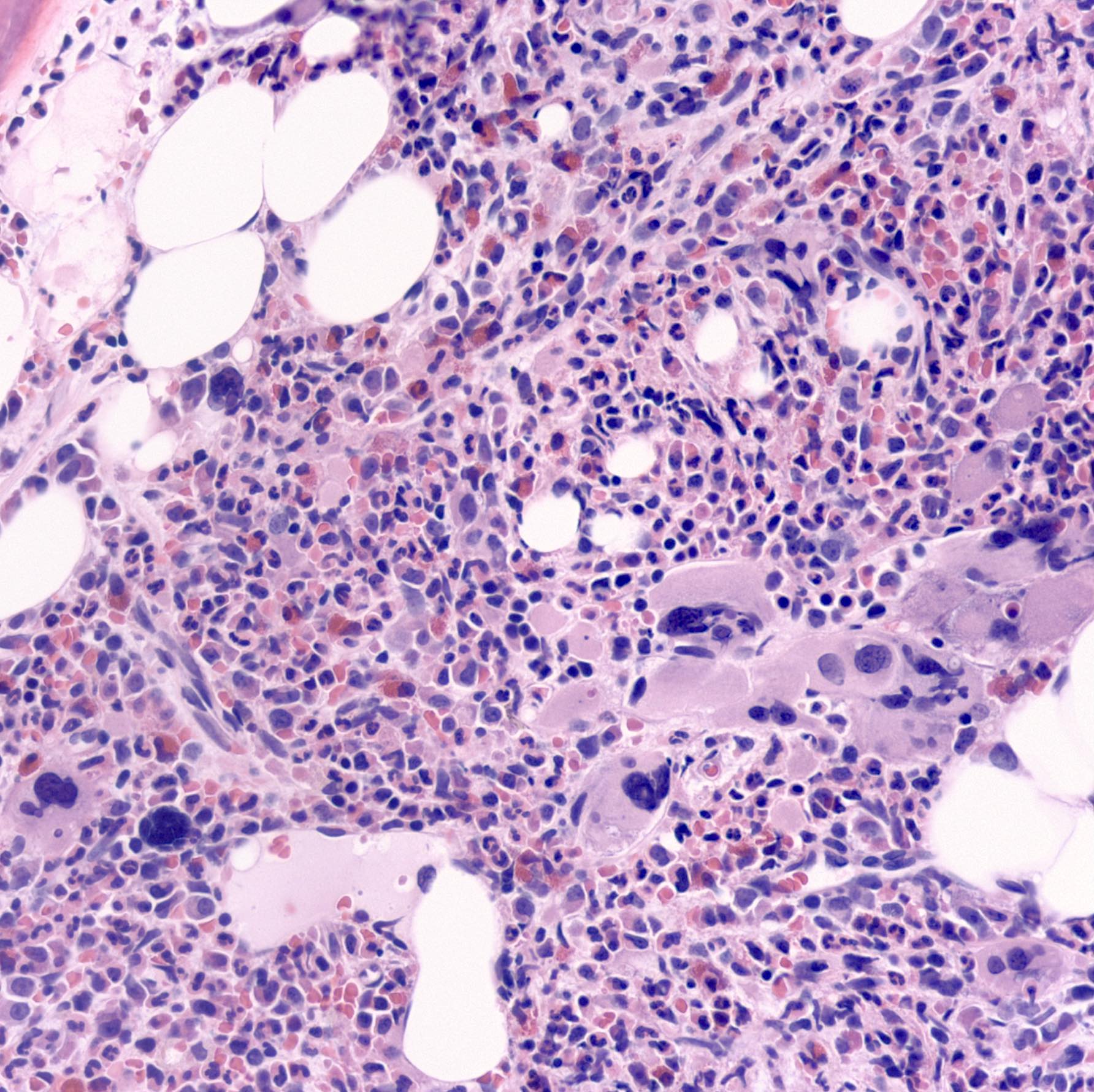
Classic finding = leukoerythroblastic blood film with “teardrop” poikilocytes. 🧐
- FBC: anaemia; early stages → raised WCC and platelets; later → pancytopenia.
- Blood film: teardrop cells, nucleated RBCs, immature granulocytes, giant platelets.
- Bone marrow: “dry tap” (aspiration failure); trephine biopsy shows fibrosis.
- Cytogenetics: no Philadelphia chromosome (distinguishes from CML).
- Raised LDH and uric acid (cell turnover).
- LAP score: normal or ↑ (low in CML).
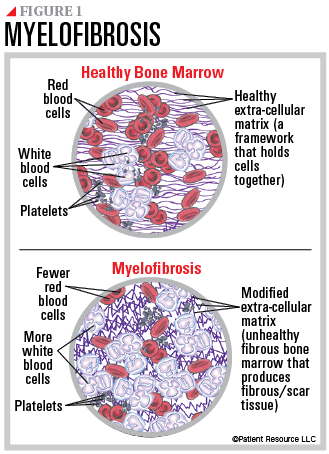
🔎 Differential: Myelofibrosis vs CML
- Myelofibrosis: marrow fibrosis, teardrop cells, splenomegaly, ↑/↓ LAP, no BCR-ABL.
- CML: marked leucocytosis, basophilia, BCR-ABL fusion gene, ↓ LAP score.
💊 Management
- Supportive: transfusions, folate, allopurinol (prevent gout).
- Hydroxycarbamide: reduces splenomegaly and blood counts.
- JAK2 inhibitors (e.g., ruxolitinib): reduce symptoms and splenomegaly.
- Splenectomy / irradiation: palliative for massive splenomegaly.
- Allogeneic stem cell transplant: only curative option; reserved for younger patients with high-risk disease.
- Median survival: ~3–5 years (longer in indolent disease, shorter in aggressive forms).
- Common causes of death: infection, haemorrhage, AML transformation.
📚 References
Cases - Myelofibrosis
- Case 1 - Splenomegaly and Cytopenia: A 68-year-old man presents with weight loss, night sweats, and early satiety. Exam: massive splenomegaly. FBC: Hb 9.0 g/dL, WCC 4.0 ×10⁹/L, platelets 80 ×10⁹/L. Blood film shows tear-drop poikilocytes (dacrocytes). Bone marrow aspirate is “dry tap”; trephine biopsy shows fibrosis. Diagnosis: Primary myelofibrosis.
- Case 2 - Post-Polycythaemia Vera Myelofibrosis: A 72-year-old woman with a 12-year history of polycythaemia vera now develops progressive anaemia, night sweats, and bone pain. Spleen is palpable 12 cm below costal margin. Bloods: pancytopenia with leukoerythroblastic film. Diagnosis: Secondary myelofibrosis following PV.
- Case 3 - Young Adult with Constitutional Symptoms: A 40-year-old man presents with severe fatigue, weight loss, and bone pain. FBC: Hb 10.5 g/dL, WCC 18 ×10⁹/L, platelets 600 ×10⁹/L. JAK2 mutation positive. Film shows tear-drop red cells, nucleated RBCs, and immature myeloid cells. Diagnosis: Myelofibrosis (proliferative phase, JAK2-positive).
Teaching Commentary 🌿
Myelofibrosis is a myeloproliferative neoplasm where clonal proliferation of haematopoietic stem cells leads to cytokine-driven bone marrow fibrosis. Blood film shows a leukoerythroblastic picture with tear-drop RBCs. Patients often have constitutional symptoms (fever, sweats, weight loss), massive splenomegaly (extramedullary haematopoiesis), and cytopenias. Mutations: JAK2, CALR, MPL. Prognosis is variable, with risk of transformation to AML. Management: - Supportive (transfusions, hydroxycarbamide for cytoreduction), - JAK inhibitors (ruxolitinib) for splenomegaly/symptoms, - Allogeneic stem cell transplantation in fit younger patients (curative option).
| The content on this website is provided for educational and informational purposes only to support exam preparation (e.g., MLA, MRCP, USMLE) and learning. This is NOT medical advice, diagnosis, treatment, or professional guidance. It does not replace consultation with a qualified healthcare professional, official guidelines (e.g., NICE, GMC, BNF), or supervised clinical practice. Always verify information with current, authoritative sources. Makindo and its contributors accept no liability for any reliance on this content, including errors, omissions, or any resulting harm, loss, or consequences. By using this site, you agree to these terms. |
|
|
Categories
- About
- Acute Medicine
- Anaesthetics and Critical Care
- Anatomy
- Anatomy and Physiology
- Biochemistry
- Book
- Cardiology
- Collections
- CompSci
- Crib Sheets
- Critical care
- Dental
- Dermatology
- Differentials
- Drugs
- ENT
- Electrocardiogram
- Embryology
- Emergency Medicine
- Endocrinology
- Ethics
- Foundation Doctors
- GCSE
- Gastroenterology
- General Practice
- Genetics
- Geriatric Medicine
- Geriatrics
- Guidelines
- Haematology
- Hepatology
- Immunology
- Infectious Diseases
- Infographic
- Investigations
- Lists
- MRCP
- Mandatory Training
- Medical Students
- Microbiology
- Nephrology
- Neurology
- Neurosurgery
- Nutrition
- OSCE
- Obstetrics Gynaecology
- Oncology
- Ophthalmology
- Oral Medicine and Dentistry
- Orthopaedics
- Paediatrics
- Palliative
- Palliative Care
- Pathology
- Pharmacology
- Physiology
- Procedures
- Psychiatry
- Public Health
- Radiology
- Respiratory
- Resuscitation
- Revision Topics
- Rheumatology
- Statistics and Research
- Stroke
- Surgery
- Toxicology
- Trauma and Orthopaedics
- USMLE
- Urology
- Vascular Surgery