
| Download the amazing global Makindo app: ✅ Means NICE/National Guidelines 2026 compliant Android | Apple | |
|---|---|
| MEDICAL DISCLAIMER: Educational use only. Not for diagnosis or management. See below for full disclaimer. |
Idiopathic Pulmonary Fibrosis
Related Subjects: |Idiopathic Pulmonary Fibrosis |Diffuse Parenchymal Lung disease |Asbestos Related Lung disease |Sarcoidosis |Coal Worker's Pneumoconiosis |Silicosis |Farmer's Lung |Cryptogenic Organising Pneumonia (COP-BOOP) |Extrinsic Allergic alveolitis (Hypersensitivity) |Byssinosis |Pneumoconiosis |Cor Pulmonale |Chest X Ray Interpretation
🫁 Idiopathic Pulmonary Fibrosis (IPF) is a chronic, progressive interstitial lung disease of unknown cause. Median survival is ~2–5 years. Always consider referral to an ILD MDT (multidisciplinary team).
📖 About
- Previously termed Cryptogenic Fibrosing Alveolitis.
- Diffuse parenchymal lung disease with relentless fibrosis of alveoli & interstitium → “honeycomb lung.”
- Rarely, an acute presentation (Hamman–Rich syndrome).
📊 Epidemiology
- Risk factors: 🚬 smoking, exposure to wood/metal dust, some viral triggers.
- More common in men, usually >50 yrs.
- Strong association with ↑ risk of lung cancer.
⚙️ Pathophysiology
- Genetic predisposition: telomerase mutations, MUC5B gene variant.
- Repeated alveolar epithelial injury → abnormal repair → fibrosis.
- Fibrogenic mediators: TNF, TGF-β, PDGF, IGF-1, endothelin-1.
- Excess fibroblast proliferation & collagen → stiff, non-compliant lungs.
🔬 Histology
- Usual Interstitial Pneumonia (UIP): Patchy fibrosis, architectural distortion, honeycomb cysts, minimal inflammation.
- Desquamative Interstitial Pneumonia (DIP): More inflammatory, alveolar macrophages, “ground glass” on HRCT, better steroid response.
🩺 Clinical Features
- Progressive exertional dyspnoea and dry cough over months.
- Velcro-like inspiratory crackles at bases.
- Finger clubbing in ~50% (similar to asbestosis).
- Weight loss, fatigue, ↓ exercise tolerance.
- Haemoptysis = 🚩 consider malignancy or PE.
- Late disease → pulmonary hypertension + cor pulmonale.
🔍 Differentials
- Occupational lung disease: 🪨 asbestosis, silicosis.
- Connective tissue diseases: Scleroderma, RA, SLE.
- Sarcoidosis, hypersensitivity pneumonitis.
- Lymphangitis carcinomatosis, miliary TB, fungal infections.
- Drug-induced (e.g., busulfan, methotrexate, amiodarone, nitrofurantoin).
🧪 Investigations
- PFTs: Restrictive pattern, ↓ FVC, ↓ TLC, ↓ DLCO.
- ABG: Resting/exercise hypoxaemia → Type 1 resp failure.
- Autoantibodies: ANA, RF positive in up to 30%.
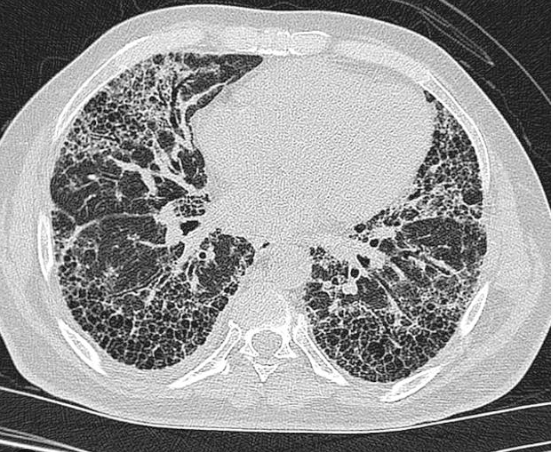
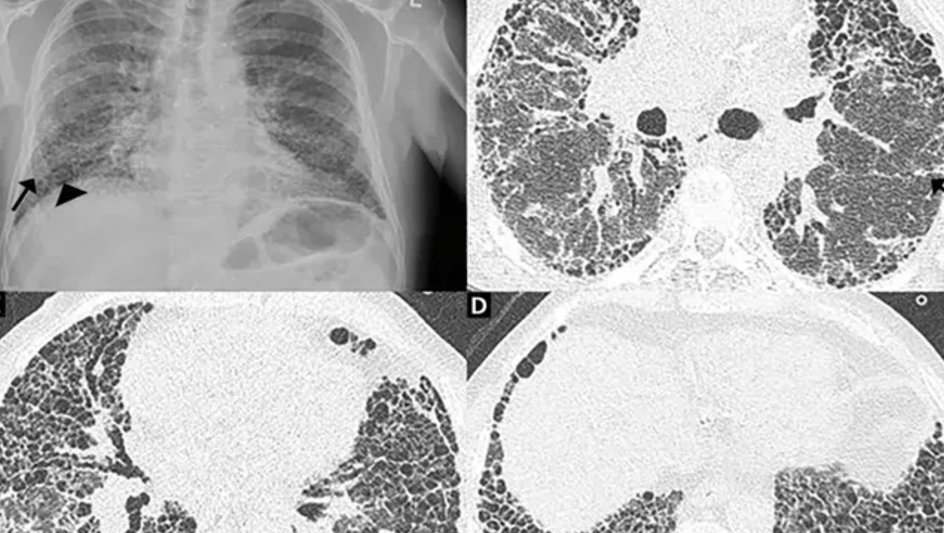
- CXR: Bilateral basal reticular shadowing.
- HRCT:
- UIP → subpleural honeycombing, traction bronchiectasis.
- DIP → ground-glass, better steroid response.
- Lung Biopsy: Consider if HRCT not diagnostic (risk of complications).
⚠️ Complications
- Progressive respiratory failure.
- Pulmonary hypertension & cor pulmonale.
- Recurrent chest infections.
- ↑ Risk of lung cancer.
- Death usually from respiratory failure.
📉 Prognostic Indicators
- Better: 👩⚕️ younger females, ground-glass on HRCT, DIP histology, steroid response.
- Poorer: Honeycombing, older age, male sex, persistent hypoxia.
💊 Management
- Antifibrotics: Pirfenidone & Nintedanib (NICE-approved if FVC 50–80% predicted).
- Lifestyle: 🚭 stop smoking, optimise nutrition, graded exercise.
- Symptom control: PPI for reflux, cough suppressants, pulmonary rehab.
- Vaccination: Pneumococcal + annual flu vaccine.
- Antibiotics: Treat infections promptly.
- Lung Transplant: Consider in suitable patients (single or double lung).
- Palliation: Oxygen therapy, opioids for dyspnoea, early palliative care input.
📚 References
3 Clinical Cases - Idiopathic Pulmonary Fibrosis (IPF) 🌫️🫁
- Case 1 - Progressive breathlessness 🚶♂️: A 68-year-old ex-smoker presents with 12 months of progressive exertional dyspnoea and a dry cough. Examination: fine “Velcro” crackles at the lung bases and digital clubbing. HRCT: basal, subpleural reticular opacities with honeycombing. Teaching: Classic presentation of IPF. Diagnosis relies on HRCT pattern (usual interstitial pneumonia, UIP). Other causes of fibrosis must be excluded. Prognosis is poor, with median survival 3–5 years.
- Case 2 - Acute exacerbation 🌡️: A 72-year-old man with known IPF presents with rapidly worsening breathlessness over 5 days. CXR: new bilateral diffuse ground-glass opacities superimposed on chronic fibrotic changes. Teaching: Acute exacerbations of IPF resemble ARDS and carry a high mortality. Often triggered by infection, surgery, or sometimes idiopathic. Management is mainly supportive (oxygen, steroids, palliative care planning).
- Case 3 - Assessment for transplant 🔄: A 55-year-old man with IPF (FVC 40%, DLCO 32%) struggles with activities of daily living. He desaturates to 82% on a 6-minute walk test. Teaching: Lung transplantation is the only treatment shown to improve survival in advanced IPF. Patients must be referred early, before severe comorbidity develops. Antifibrotic agents (pirfenidone, nintedanib) may slow decline but do not cure.
| The content on this website is provided for educational and informational purposes only to support exam preparation (e.g., MLA, MRCP, USMLE) and learning. This is NOT medical advice, diagnosis, treatment, or professional guidance. It does not replace consultation with a qualified healthcare professional, official guidelines (e.g., NICE, GMC, BNF), or supervised clinical practice. Always verify information with current, authoritative sources. Makindo and its contributors accept no liability for any reliance on this content, including errors, omissions, or any resulting harm, loss, or consequences. By using this site, you agree to these terms. |
|
|
Categories
- About
- Acute Medicine
- Anaesthetics and Critical Care
- Anatomy
- Anatomy and Physiology
- Biochemistry
- Book
- Cardiology
- Collections
- CompSci
- Crib Sheets
- Critical care
- Dental
- Dermatology
- Differentials
- Drugs
- ENT
- Electrocardiogram
- Embryology
- Emergency Medicine
- Endocrinology
- Ethics
- Foundation Doctors
- GCSE
- Gastroenterology
- General Practice
- Genetics
- Geriatric Medicine
- Geriatrics
- Guidelines
- Haematology
- Hepatology
- Immunology
- Infectious Diseases
- Infographic
- Investigations
- Lists
- MRCP
- Mandatory Training
- Medical Students
- Microbiology
- Nephrology
- Neurology
- Neurosurgery
- Nutrition
- OSCE
- Obstetrics Gynaecology
- Oncology
- Ophthalmology
- Oral Medicine and Dentistry
- Orthopaedics
- Paediatrics
- Palliative
- Palliative Care
- Pathology
- Pharmacology
- Physiology
- Procedures
- Psychiatry
- Public Health
- Radiology
- Respiratory
- Resuscitation
- Revision Topics
- Rheumatology
- Statistics and Research
- Stroke
- Surgery
- Toxicology
- Trauma and Orthopaedics
- USMLE
- Urology
- Vascular Surgery