
| Download the amazing global Makindo app: ✅ Means NICE/National Guidelines 2026 compliant Android | Apple | |
|---|---|
| MEDICAL DISCLAIMER: Educational use only. Not for diagnosis or management. See below for full disclaimer. |
Hereditary Haemorrhagic Telangiectasia (HHT) ✅
Related Subjects: |Iron deficiency Anaemia |Haemolytic anaemia |Macrocytic anaemia |Megaloblastic anaemia |Microcytic anaemia |Myelodysplasia |Myelofibrosis |Hereditary Spherocytosis |Hereditary Elliptocytosis |Haemophilia A |Haemophilia B |Haemolytic anaemia |Heme |Globins |Red blood cells |White blood cells |Lymphocytes |Platelets |Cryoprecipitate |Fresh Frozen Plasma |Blood Cell Maturation |Blood film interpretation |Reticulocytes
🧬 Hereditary Haemorrhagic Telangiectasia (HHT), also called Osler–Weber–Rendu disease, is a rare autosomal dominant vascular disorder. It causes fragile abnormal blood vessels - telangiectasia and arteriovenous malformations (AVMs) - that can bleed or shunt blood abnormally. 🚨 Early recognition matters because complications include iron deficiency anaemia, haemorrhagic stroke, paradoxical embolic stroke, and brain abscess.
📖 About
- Definition: inherited disorder of abnormal vascular development causing telangiectasia and AVMs.
- Inheritance 👪: autosomal dominant.
- Commonly affected sites: 👃 nose, 👄 lips, 👅 tongue, 🫁 lungs, 🍽️ GI tract, 🧠 brain, and sometimes 🫀 liver.
- Family history: many affected patients have a first-degree relative with HHT.
- Pulmonary AVMs 🫁: important because they can allow paradoxical emboli and cause hypoxaemia.
- Brain vascular malformations 🧠: clinically important because they may bleed.
🔎 Think of HHT in a patient with recurrent epistaxis + iron deficiency anaemia + mucocutaneous telangiectasia + family history. 💡 Once suspected, do not stop at the nose - screen for visceral AVMs, especially in the lungs.
🧬 Aetiology
- Disorder of angiogenesis and vascular remodelling.
- Genes: ENG (HHT1), ACVRL1 (HHT2), and SMAD4 (HHT with juvenile polyposis overlap).
- These mutations affect signalling involved in normal vessel maturation, so fragile direct artery–vein connections form without a normal capillary bed.
🧠 Why HHT causes stroke
- 💥 Haemorrhagic stroke: rupture of a cerebral vascular malformation.
- 🫁➡️🧠 Paradoxical embolic stroke: thrombus bypasses the lung filter through a pulmonary AVM and reaches the brain.
- 🦠 Brain abscess: septic emboli can also pass through pulmonary AVMs.
⚡ Clinical Features
- 👃 Epistaxis: recurrent spontaneous nosebleeds - often the earliest and commonest feature.
- 🩸 Iron deficiency anaemia: chronic blood loss from epistaxis or GI telangiectasia.
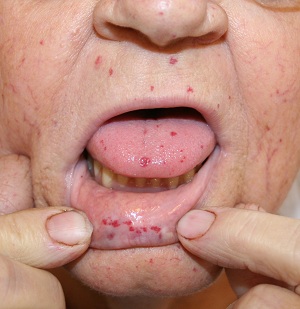
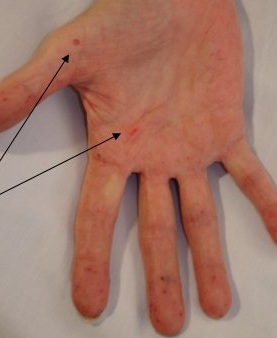
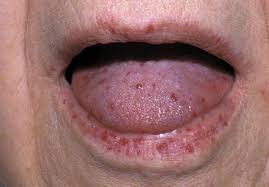
- 👄 Mucocutaneous telangiectasia: lips, oral cavity, tongue, fingertips, face, and nasal mucosa.
- 🫁 Pulmonary AVMs: dyspnoea, hypoxaemia, haemoptysis, paradoxical emboli.
- 🍽️ GI bleeding: melaena, occult blood loss, or chronic anaemia, especially in older adults.
- 🧠 Neurological complications: stroke, seizures, headache, brain abscess, or haemorrhage.
- 🫀 Hepatic involvement: may cause high-output cardiac failure in some patients.
🖼️ Suggested Extra Images to Add
- 🫁 CT image of a pulmonary AVM
- 🧠 MRI/angiographic image of a cerebral AVM
- 👃 Nasal telangiectasia / epistaxis photograph
- 🧬 Simple inheritance diagram showing autosomal dominant transmission
📊 Diagnostic Criteria (Curaçao Criteria)
| Criterion | Description |
|---|---|
| 👃 Epistaxis | Spontaneous, recurrent nosebleeds. |
| 🔴 Telangiectasia | Characteristic lesions on lips, oral cavity, fingers, or nose. |
| 🫁🧠🍽️ Visceral lesions | Pulmonary, cerebral, hepatic, GI, or spinal AVMs/telangiectasia. |
| 👪 Family history | First-degree relative with confirmed HHT. |
✅ Definite HHT: 3 or more criteria
⚠️ Possible HHT: 2 criteria
❌ Unlikely HHT: fewer than 2 criteria
🔬 Investigations
- 🩸 Bloods: FBC, ferritin, iron studies, B12/folate, U&Es, LFTs.
- 🫁 Pulmonary AVM screening: bubble echo / transthoracic contrast echocardiography is the preferred initial screening test.
- 🫁 CT chest: used to define pulmonary AVMs for treatment planning if screening is positive.
- 🧠 MRI brain: used in specialist pathways to assess for brain vascular malformations.
- 🍽️ GI investigation: endoscopy / capsule endoscopy if iron deficiency anaemia or overt bleeding suggests GI involvement.
- 🧬 Genetic testing: helps confirm subtype and guide family screening.
🩺 Management
- 🚑 Emergency care: ABC approach, resuscitation, blood transfusion if needed, and urgent specialist input for major haemorrhage or neurological events.
- 👃 Epistaxis: humidification, topical measures, iron replacement, ENT review; refractory cases may need cautery or laser therapy.
- 🩸 Iron deficiency / GI bleeding: oral or IV iron, endoscopic therapy where appropriate, and transfusion if severe.
- 🫁 Pulmonary AVMs: endovascular embolisation is standard treatment for suitable lesions.
- 💊 Antifibrinolytics: tranexamic acid may help troublesome bleeding in selected cases.
- ⚖️ Antithrombotics: not absolutely contraindicated, but require careful individual bleeding/thrombotic risk assessment.
- 👪 Genetics: family counselling and cascade screening are important.
🫁 Important practical pearl: Pulmonary AVMs can let clots and bacteria bypass the lung filter. This explains why HHT can cause embolic stroke and brain abscess even in relatively young patients.
📈 Prognosis
- 📆 Chronic but highly variable disease course.
- ✅ Many patients live well with good supportive care and surveillance.
- ⚠️ Major complications include chronic anaemia, pulmonary AVM complications, stroke, and haemorrhage.
- 🌱 Early identification and treatment of AVMs improves long-term outcomes.
🧠 Teaching Commentary
HHT is best understood as a disorder of vascular architecture: instead of a normal artery → capillary → vein network, patients develop fragile direct vascular connections. That explains both the easy bleeding of telangiectasia and the right-to-left shunting of pulmonary AVMs. Clinically, the highest-yield pattern is recurrent epistaxis + iron deficiency anaemia + telangiectasia + family history. Once suspected, always think beyond ENT disease and actively consider lung and brain complications.
📚 References
- NCBI Bookshelf – Hereditary Hemorrhagic Telangiectasia
- Systematic Review – Brain AVMs in HHT
- Treatment of AVMs
🧪 Cases – Hereditary Haemorrhagic Telangiectasia
- Case 1 👃: A 28-year-old woman has recurrent nosebleeds since childhood, iron deficiency anaemia, and red blanching lesions on her lips and tongue. Her mother had similar symptoms. Likely HHT with recurrent epistaxis and mucocutaneous telangiectasia.
- Case 2 🫁: A 40-year-old man presents with exertional dyspnoea and clubbing. CT chest shows a pulmonary AVM; contrast echo confirms right-to-left shunt. Likely HHT with pulmonary AVM causing hypoxaemia.
- Case 3 🍽️: A 65-year-old woman presents with melaena and iron deficiency anaemia. Endoscopy shows multiple gastric telangiectatic lesions. Likely HHT with GI telangiectasia causing chronic blood loss.
📸 Clinical Images
Mucocutaneous telangiectasia in HHT:
| The content on this website is provided for educational and informational purposes only to support exam preparation (e.g., MLA, MRCP, USMLE) and learning. This is NOT medical advice, diagnosis, treatment, or professional guidance. It does not replace consultation with a qualified healthcare professional, official guidelines (e.g., NICE, GMC, BNF), or supervised clinical practice. Always verify information with current, authoritative sources. Makindo and its contributors accept no liability for any reliance on this content, including errors, omissions, or any resulting harm, loss, or consequences. By using this site, you agree to these terms. |
|
|
Categories
- About
- Acute Medicine
- Anaesthetics and Critical Care
- Anatomy
- Anatomy and Physiology
- Biochemistry
- Book
- Cardiology
- Collections
- CompSci
- Crib Sheets
- Critical care
- Dental
- Dermatology
- Differentials
- Drugs
- ENT
- Electrocardiogram
- Embryology
- Emergency Medicine
- Endocrinology
- Ethics
- Foundation Doctors
- GCSE
- Gastroenterology
- General Practice
- Genetics
- Geriatric Medicine
- Geriatrics
- Guidelines
- Haematology
- Hepatology
- Immunology
- Infectious Diseases
- Infographic
- Investigations
- Lists
- MRCP
- Mandatory Training
- Medical Students
- Microbiology
- Nephrology
- Neurology
- Neurosurgery
- Nutrition
- OSCE
- Obstetrics Gynaecology
- Oncology
- Ophthalmology
- Oral Medicine and Dentistry
- Orthopaedics
- Paediatrics
- Palliative
- Palliative Care
- Pathology
- Pharmacology
- Physiology
- Procedures
- Psychiatry
- Public Health
- Radiology
- Respiratory
- Resuscitation
- Revision Topics
- Rheumatology
- Statistics and Research
- Stroke
- Surgery
- Toxicology
- Trauma and Orthopaedics
- USMLE
- Urology
- Vascular Surgery