
| Download the amazing global Makindo app: ✅ Means NICE/National Guidelines 2026 compliant Android | Apple | |
|---|---|
| MEDICAL DISCLAIMER: Educational use only. Not for diagnosis or management. See below for full disclaimer. |
Huntingtons Disease/Chorea
Related Subjects: |Dementia |Alzheimer Disease |Vascular Dementia |Frontotemporal dementia |Huntington's Disease/Chorea |Dementia with Lewy bodies |Creutzfeldt Jakob disease |Behavioural and Psychological Symptoms of Dementia
The disease is named after George Huntington, who described it in 1872 after observing families in Long Island, New York. Tracing back to settlers from Suffolk, England in the 1600s, it became one of the best-known examples of an inherited neurodegenerative disorder. If interested, see Ian McEwan’s novel "Saturday", which features a character with Huntington’s chorea. 📚
About 🧠
- Autosomal dominant inheritance – only one affected copy is needed.
- Choreiform movements 💃 usually start at age 30–50.
- Cognitive decline (executive dysfunction, difficulty multitasking) is often more disabling than the motor features.
- Family history is usually positive.
- Prevalence: ~5 per 100,000 population in Europe/North America.
Aetiology 🔬
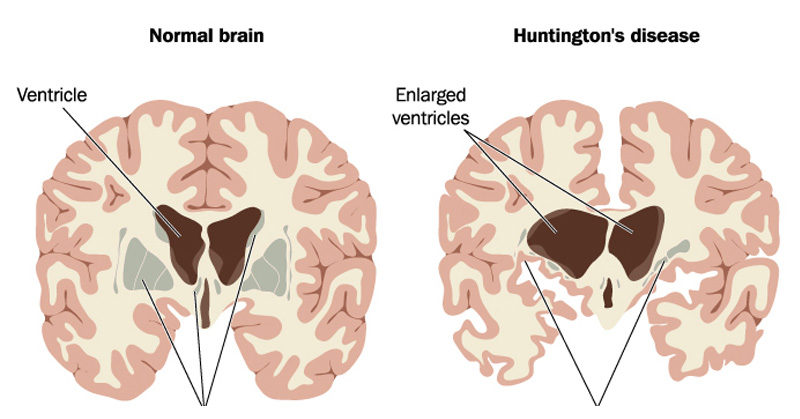
- Mutation in the HTT gene on chromosome 4p16.3.
- Expanded CAG trinucleotide repeat (>42 repeats) → abnormal huntingtin protein → neuronal toxicity.
- CAG codes for glutamine → long polyglutamine tracts → misfolded proteins and cell death.
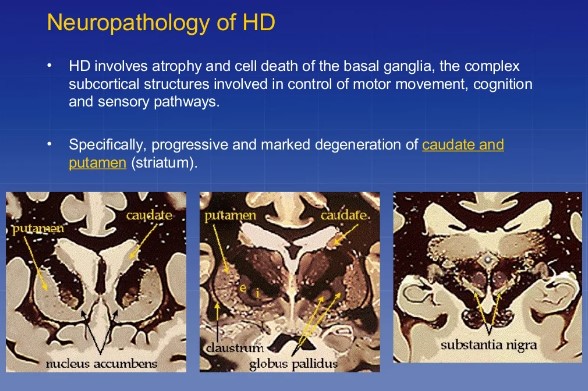
- Mainly affects the caudate and putamen (basal ganglia).
- Paternal inheritance → earlier onset (“anticipation”), sometimes juvenile Parkinsonian-like variant (Westphal variant).
The juvenile form (onset <21 years) presents with rigidity, spasticity, dystonia, and seizures rather than chorea.
Clinical Features ⚕️
- Motor: Restlessness → chorea → later rigidity, spasticity, dystonia. Increased blink rate, slow eye movements, balance problems.
- Cognitive: Subcortical dementia – poor organisation, planning, prioritisation, slowed thought.
- Psychiatric: Depression, anxiety, irritability, episodic anger, OCD-like behaviours, and sometimes psychosis.
- Other: Dysphagia (late stage), epilepsy, weight loss, falls.
- Anticipation: Earlier and more severe in successive generations.
Investigations 🧪
- CT/MRI: Caudate atrophy → enlarged lateral ventricles (“boxcar ventricles”).
- PET scan: Early ↓ metabolism in caudate before structural atrophy.
- Genetic testing: Detects CAG expansion – available via regional genetics clinics (ethical implications, predictive testing in asymptomatic relatives).
- Pathology: Atrophy of caudate, putamen, and cortex; neuronal loss with gliosis.
Differentials 🔍
- Sydenham’s chorea (post-strep, self-limiting).
- Wilson’s disease (younger age, hepatic involvement, Kayser-Fleischer rings).
Management 💊
- Supportive: Multidisciplinary team, physiotherapy, OT, speech therapy, genetic counselling for family.
- Chorea control: Tetrabenazine (dopamine depleting), Haloperidol, or Sulpiride – ⚠️ can worsen parkinsonism/depression.
- Psychiatric symptoms: SSRIs for depression; mood stabilisers (Valproate, Carbamazepine) for irritability/impulsivity.
- End-stage: Palliative care planning is essential as progression is inevitable.
Key Clinical Pearls ✨
- Look for chorea + cognitive decline + psychiatric changes in a middle-aged patient with family history.
- Increased blink rate is a subtle sign (opposite of Parkinson’s).
- Juvenile onset = rigidity > chorea (Westphal variant).
References 📚
Cases - Huntington’s Disease (Chorea)
- Case 1 - Early Psychiatric and Motor Features 🧩: A 38-year-old man presents with irritability, depression, and difficulty concentrating at work. His wife has noticed involuntary jerky movements of his hands and face. Family history: father died in his 50s with a “neurological illness.” Diagnosis: Early Huntington’s disease. Management: Genetic confirmation; symptomatic therapy (tetrabenazine/deutetrabenazine for chorea, SSRIs for depression); genetic counselling for family.
- Case 2 - Progressive Chorea and Cognitive Decline ⚡: A 45-year-old woman presents with worsening fidgety limb movements and difficulty walking. Exam: generalized chorea, impaired saccadic eye movements, and executive dysfunction on cognitive testing. Diagnosis: Mid-stage Huntington’s disease with cognitive impairment. Management: MDT support (neurology, psychiatry, physio, OT); dopamine-blocking drugs (e.g., risperidone) if severe chorea/psychosis.
- Case 3 - Advanced Disease and Care Needs 🛏️: A 55-year-old man with known Huntington’s is admitted from a care home with profound weight loss, rigidity, and immobility. He has dysphagia and recurrent aspiration pneumonia. Diagnosis: Advanced Huntington’s disease. Management: Palliative care focus - nutrition support, swallowing assessment, respiratory management, family support.
Teaching Commentary 🧠
Huntington’s disease is an autosomal dominant trinucleotide repeat disorder (CAG expansion, chromosome 4). Pathology: progressive degeneration of striatal neurons. Key features: - Motor: chorea, dystonia, later rigidity and bradykinesia. - Psychiatric: depression, irritability, psychosis. - Cognitive: progressive dementia, impaired executive function. Onset: typically 30s–50s, but juvenile cases exist. Investigations: genetic testing confirms diagnosis; MRI shows caudate atrophy. Management is supportive - no cure. Symptom control (VMAT inhibitors for chorea, antipsychotics for behaviour), MDT rehabilitation, genetic counselling. Prognosis: 15–20 years after onset, often death from aspiration or infection.
| The content on this website is provided for educational and informational purposes only to support exam preparation (e.g., MLA, MRCP, USMLE) and learning. This is NOT medical advice, diagnosis, treatment, or professional guidance. It does not replace consultation with a qualified healthcare professional, official guidelines (e.g., NICE, GMC, BNF), or supervised clinical practice. Always verify information with current, authoritative sources. Makindo and its contributors accept no liability for any reliance on this content, including errors, omissions, or any resulting harm, loss, or consequences. By using this site, you agree to these terms. |
|
|
Categories
- About
- Acute Medicine
- Anaesthetics and Critical Care
- Anatomy
- Anatomy and Physiology
- Biochemistry
- Book
- Cardiology
- Collections
- CompSci
- Crib Sheets
- Critical care
- Dental
- Dermatology
- Differentials
- Drugs
- ENT
- Electrocardiogram
- Embryology
- Emergency Medicine
- Endocrinology
- Ethics
- Foundation Doctors
- GCSE
- Gastroenterology
- General Practice
- Genetics
- Geriatric Medicine
- Geriatrics
- Guidelines
- Haematology
- Hepatology
- Immunology
- Infectious Diseases
- Infographic
- Investigations
- Lists
- MRCP
- Mandatory Training
- Medical Students
- Microbiology
- Nephrology
- Neurology
- Neurosurgery
- Nutrition
- OSCE
- Obstetrics Gynaecology
- Oncology
- Ophthalmology
- Oral Medicine and Dentistry
- Orthopaedics
- Paediatrics
- Palliative
- Palliative Care
- Pathology
- Pharmacology
- Physiology
- Procedures
- Psychiatry
- Public Health
- Radiology
- Respiratory
- Resuscitation
- Revision Topics
- Rheumatology
- Statistics and Research
- Stroke
- Surgery
- Toxicology
- Trauma and Orthopaedics
- USMLE
- Urology
- Vascular Surgery