Retinitis pigmentosa
Retinitis Pigmentosa (RP) is a group of inherited retinal degenerations causing progressive loss of photoreceptor cells. Patients first develop night blindness, then tunnel vision, and in advanced stages, complete blindness. When combined with hearing loss, it is known as Usher Syndrome. Early recognition and genetic testing are crucial for counselling and emerging therapies.
ℹ️ About
- Group of genetic disorders causing retinal degeneration.
- Rods affected first → night blindness, then cones → central vision loss.
- Inheritance: Autosomal recessive (most common), autosomal dominant, or X-linked.
- Progression varies widely depending on the underlying gene mutation.
🔬 Aetiology
- Caused by >60 gene mutations (e.g., rhodopsin, RPE65, USH2A).
- Disrupts photoreceptor metabolism and retinal pigment epithelium (RPE) function.
- Rod cells degenerate first → nyctalopia (night blindness).
- Some cases sporadic; male predominance due to X-linked forms.
🧬 Conditions Associated with RP
- Usher syndrome: RP + congenital or progressive deafness.
- Bardet-Biedl syndrome: RP + obesity, polydactyly, renal disease, learning disability.
- Refsum disease: RP + neuropathy, ataxia, anosmia (due to phytanic acid accumulation).
- Abetalipoproteinemia: RP + neuropathy, fat malabsorption, ataxia.
- Kearns-Sayre syndrome: RP + cardiac conduction block + external ophthalmoplegia.
- Alström syndrome: RP + obesity, diabetes, hearing loss.
🩺 Clinical Features
- Night Blindness (Nyctalopia): Early symptom due to rod dysfunction.
- Peripheral vision loss → Tunnel vision: Progressive field constriction.
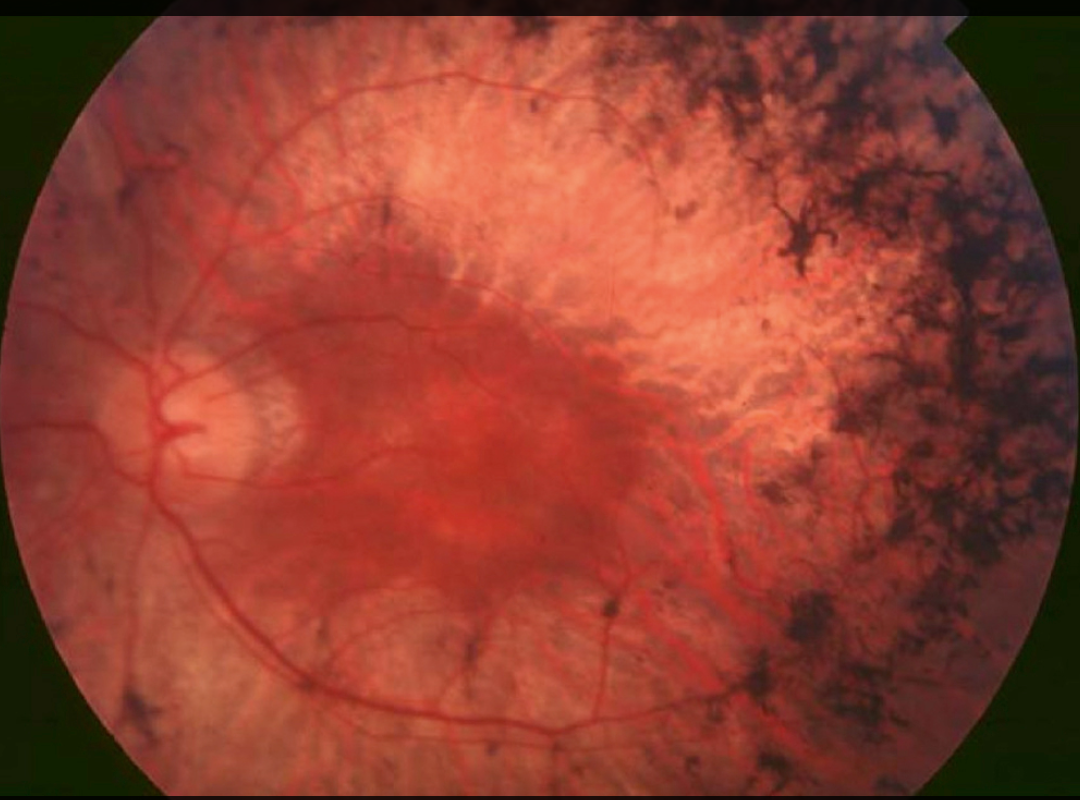
- Fundus exam: Bone spicule pigmentation, attenuated arterioles, optic disc pallor.
- Photopsia: Flashes of light in dim environments.
- Gradual progression → eventual central vision loss in advanced stages.
🔎 Investigations
- Electroretinogram (ERG): Reduced/absent rod and cone responses (diagnostic).
- Genetic testing: Confirms mutation, informs prognosis, family planning, and gene therapy eligibility.
- Visual field testing: Maps tunnel vision progression.
- OCT: Retinal thinning, photoreceptor loss.
- Fundus photography: Documents bony spicule pigmentation & optic disc changes.
💊 Management
- Vitamin A palmitate: 15,000 IU/day may slow progression (monitor for toxicity).
- Gene therapy: Luxturna (voretigene neparvovec) for RPE65 mutations approved in some centres.
- Low vision aids: Magnifiers, screen readers, mobility training.
- Retinal prostheses: Argus II (“bionic eye”) under evaluation.
- Genetic counselling: For patients and families regarding inheritance.
- Supportive: Mobility training, guide dogs, counselling.
📊 Comparison: RP vs Other Retinal Degenerations
| Feature | Retinitis Pigmentosa | Macular Degeneration | Diabetic Retinopathy |
|---|
| Onset | Childhood–early adulthood | Late adulthood | Variable (linked to diabetes) |
| Initial Symptom | Night blindness | Central vision loss | Blurring, floaters |
| Vision Loss Pattern | Peripheral → Tunnel vision → Central | Central first | Patchy, variable |
| Fundus Findings | Bone spicules, pale disc, narrow vessels | Drusen, pigment changes | Microaneurysms, hemorrhages, exudates |
📈 Prognosis
- Progressive vision loss over decades.
- Peripheral vision lost first; central vision retained until later life.
- Some retain functional vision into mid-life; others progress rapidly depending on mutation.
🔑 Clinical Pearls
- Always ask about hearing loss → Usher syndrome.
- Bone spicule pigmentation on fundoscopy is diagnostic.
- Vitamin A should only be started under ophthalmologist guidance (risk of hepatotoxicity).
- Gene therapy is revolutionising care in specific subtypes (e.g., RPE65).
References
Cases - Retinitis Pigmentosa (RP)
- Case 1 - Night blindness 🌙: A 14-year-old boy presents with difficulty seeing in dim light and bumping into objects at night. Family history: father affected. Fundoscopy: bone spicule pigmentation, attenuated vessels, pale optic disc. Diagnosis: retinitis pigmentosa (autosomal dominant). Managed with low-vision aids, genetic counselling, and vitamin A supplementation under specialist advice.
- Case 2 - Peripheral field loss 🔭: A 22-year-old woman complains of tunnel vision affecting her ability to drive. Visual field testing: concentric peripheral field loss. Fundus: classic RP changes. Diagnosis: RP with progressive visual field constriction. Managed with occupational therapy, referral to DVLA for driving standards, and genetic testing for future family planning.
- Case 3 - Syndromic RP 🧬: A 10-year-old boy with obesity, polydactyly, and learning difficulties presents with night blindness. Fundoscopy: pigmentary retinopathy. Genetic testing: Bardet–Biedl syndrome. Diagnosis: syndromic RP. Managed with low-vision support, genetic counselling, and multidisciplinary care (endocrinology, nephrology).
Teaching Point 🩺: Retinitis pigmentosa is a group of inherited retinal dystrophies causing progressive rod-cone degeneration.
Key features: night blindness, peripheral field loss (tunnel vision), bone spicule pigmentation.
Associations: cataracts, keratoconus, syndromes (Bardet–Biedl, Usher).
Management: no cure; supportive with low-vision aids, genetic counselling, and emerging gene/retinal therapies.
