
| Download the amazing global Makindo app: ✅ Means NICE/National Guidelines 2026 compliant Android | Apple | |
|---|---|
| MEDICAL DISCLAIMER: Educational use only. Not for diagnosis or management. See below for full disclaimer. |
Fabry disease
Related Subjects: |Autosomal Dominant |Autosomal Recessive |X Linked Recessive |Osteoporosis
🧬 Fabry disease is a rare X-linked lysosomal storage disorder caused by deficiency of the enzyme α-galactosidase A. This leads to pathological accumulation of globotriaosylceramide (GL-3 or Gb3) in multiple organs. The result is a multi-systemic disease with dermatological, renal, neurological, ocular, and cardiovascular involvement. Without treatment, progressive organ failure shortens life expectancy.
📖 About
- Inherited in an X-linked recessive manner. Males are usually more severely affected, but heterozygous females can also show symptoms due to random X-inactivation (lyonisation).
- Incidence: ~1 in 40,000 males 👶, though newborn screening suggests higher prevalence of late-onset variants.
- One of the few lysosomal storage diseases for which enzyme replacement therapy (ERT) is available.
- Classic Fabry disease often presents in childhood; late-onset forms may present only with cardiac or renal disease in adulthood.
🧬 Aetiology & Pathophysiology
- Mutations in the GLA gene (Xq22.1) → deficiency of α-galactosidase A enzyme.
- Accumulation of GL-3 in lysosomes of vascular endothelial cells, smooth muscle, renal epithelial cells, and cardiac myocytes → progressive cellular dysfunction.
- Vascular GL-3 deposition → impaired blood flow, inflammation, and fibrosis → multi-organ complications (stroke, cardiomyopathy, renal failure).
- Blood group B individuals may be more severely affected since α-GAL A also helps break down B antigens.
🌈 Clinical Features
- Skin 🟤: Angiokeratomas (tiny, dark red-purple skin lesions), classically in a "bathing trunk" distribution (between knees and umbilicus).
- Neurological ⚡: Pain crises (acroparaesthesiae) with burning pain in hands/feet, especially in childhood; risk of early strokes (often posterior circulation).
- Renal 💧: Proteinuria, polyuria, progressive CKD; may progress to ESRD. Fanconi-like tubular dysfunction possible.
- Cardiac ❤️: LVH, arrhythmias, heart failure, valvular disease → major cause of death in late-onset forms.
- Ocular 👁️: Cornea verticillata (whorled corneal deposits), lens opacities, vascular tortuosity.
- Other: Hypohidrosis (↓ sweating), heat intolerance, lymphoedema, osteoporosis, GI symptoms (diarrhoea, abdominal pain).
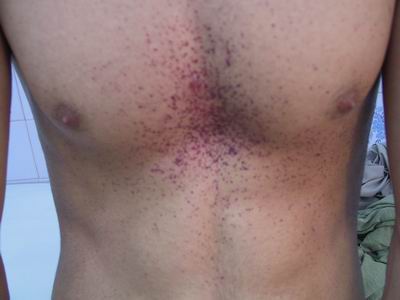
Fabry disease: GL-3 accumulation causes systemic involvement
🔍 Investigations
- Enzyme Assay 🧪: Low α-galactosidase A activity in leukocytes, plasma, or dried blood spot confirms diagnosis in males.
- Genetic Testing 🧬: Identifies GLA mutation; crucial for confirming carrier status in females.
- Urine Analysis: Proteinuria + elevated GL-3.
- MRI Brain 🧠: May show white matter lesions and pulvinar sign (posterior thalamic calcification).
- Cardiac Work-up: ECG/echo for LVH, arrhythmias; cardiac MRI can show late gadolinium enhancement (fibrosis).
- Renal Biopsy: Zebra bodies on EM; reserved for atypical cases.
- Ophthalmology 👁️: Slit-lamp reveals cornea verticillata.
- Prenatal Diagnosis: Enzyme assay in amniocytes or chorionic villus sampling.
💊 Management
- Enzyme Replacement Therapy (ERT) 💉: Recombinant α-galactosidase A (agalsidase alfa or beta) slows GL-3 accumulation and organ damage.
- Chaperone Therapy 💊: Migalastat stabilises certain mutant enzymes (only in amenable mutations).
- Renal Care 💧: ACE inhibitors/ARBs for proteinuria, dialysis or renal transplantation for ESRD.
- Cardiac Care ❤️: Arrhythmia monitoring, pacemakers, heart failure therapy, cardiac MRI surveillance.
- Pain Control ⚡: Neuropathic agents (gabapentin, carbamazepine, TCAs).
- Supportive: Physical therapy, psychological support, patient education, and genetic counselling for families.
📌 Clinical Pearls
- Classic male Fabry = angiokeratomas + neuropathic pain + corneal verticillata + renal dysfunction.
- Females can be anything from asymptomatic carriers to as severe as males.
- Always consider in young patients with stroke (esp. posterior circulation, LVH, proteinuria).
- ERT works best when started before irreversible organ damage has occurred.
📚 References
| The content on this website is provided for educational and informational purposes only to support exam preparation (e.g., MLA, MRCP, USMLE) and learning. This is NOT medical advice, diagnosis, treatment, or professional guidance. It does not replace consultation with a qualified healthcare professional, official guidelines (e.g., NICE, GMC, BNF), or supervised clinical practice. Always verify information with current, authoritative sources. Makindo and its contributors accept no liability for any reliance on this content, including errors, omissions, or any resulting harm, loss, or consequences. By using this site, you agree to these terms. |
|
|
Categories
- About
- Acute Medicine
- Anaesthetics and Critical Care
- Anatomy
- Anatomy and Physiology
- Biochemistry
- Book
- Cardiology
- Collections
- CompSci
- Crib Sheets
- Critical care
- Dental
- Dermatology
- Differentials
- Drugs
- ENT
- Electrocardiogram
- Embryology
- Emergency Medicine
- Endocrinology
- Ethics
- Foundation Doctors
- GCSE
- Gastroenterology
- General Practice
- Genetics
- Geriatric Medicine
- Geriatrics
- Guidelines
- Haematology
- Hepatology
- Immunology
- Infectious Diseases
- Infographic
- Investigations
- Lists
- MRCP
- Mandatory Training
- Medical Students
- Microbiology
- Nephrology
- Neurology
- Neurosurgery
- Nutrition
- OSCE
- Obstetrics Gynaecology
- Oncology
- Ophthalmology
- Oral Medicine and Dentistry
- Orthopaedics
- Paediatrics
- Palliative
- Palliative Care
- Pathology
- Pharmacology
- Physiology
- Procedures
- Psychiatry
- Public Health
- Radiology
- Respiratory
- Resuscitation
- Revision Topics
- Rheumatology
- Statistics and Research
- Stroke
- Surgery
- Toxicology
- Trauma and Orthopaedics
- USMLE
- Urology
- Vascular Surgery