
| Download the amazing global Makindo app: ✅ Means NICE/National Guidelines 2026 compliant Android | Apple | |
|---|---|
| MEDICAL DISCLAIMER: Educational use only. Not for diagnosis or management. See below for full disclaimer. |
Neurofibromatosis Type 1
Related Subjects: |Neurofibromatosis Type 1 |Neurofibromatosis Type 2 |Tuberous sclerosis |Café-au-lait spots |McCune Albright syndrome
📊 About
- Prevalence: NF1 is around 10× more common than NF2. Estimated prevalence ≈ 1 in 3,000. 🧬
- Exam Focus: NF1 features are far more likely to appear in exams than NF2 due to its distinctive skin, eye, and tumour manifestations. 🎓
🧬 Aetiology
- Inheritance: Autosomal dominant (50% chance of transmission), but ~50% arise from de novo mutations. 👪
- Gene: Mutation in NF1 gene on chromosome 17, encoding neurofibromin, a tumour suppressor protein regulating the Ras/MAPK pathway.
- Pathophysiology: Loss of neurofibromin → ↑ Ras activity → uncontrolled cell growth → neurofibroma formation. 🔬
🩺 Clinical Features
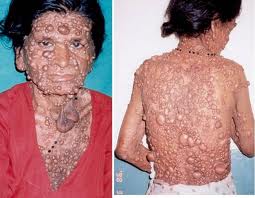
- Cutaneous Neurofibromas: Multiple, soft, rubbery nodules under the skin. May cause cosmetic concerns or compress structures.
- Plexiform Neurofibromas: Larger, deeper tumours, can cause disfigurement or functional loss; risk of malignant peripheral nerve sheath tumour (~10%). ⚠️
- Axillary/Inguinal Freckling: “Crowe’s sign” – pathognomonic for NF1. 🌟
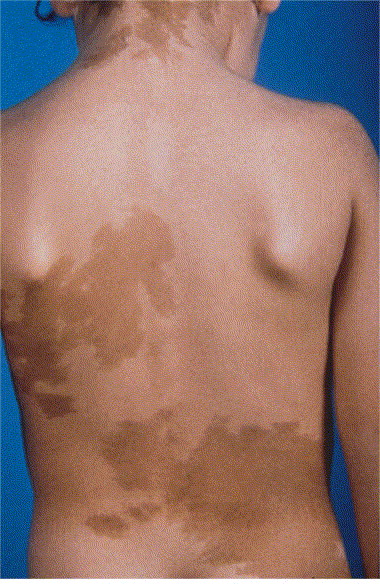
- Café-au-lait Spots: ≥6 macules (>5 mm in children, >15 mm in adults). Best seen with Wood’s lamp on pale skin. ☕
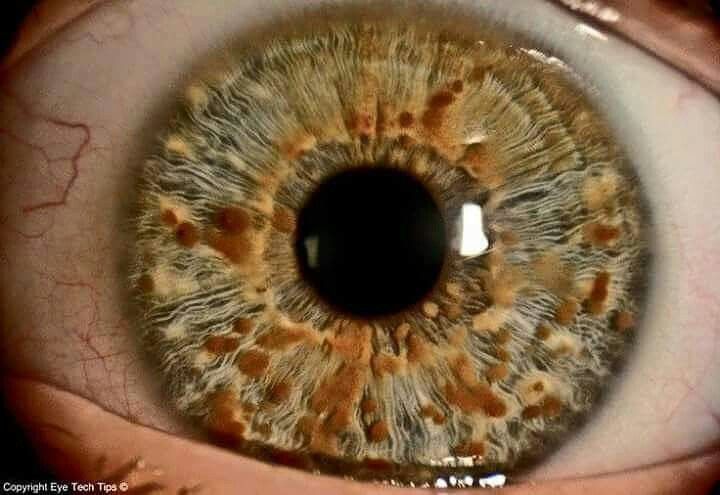
- Lisch Nodules: Iris hamartomas, visible on slit-lamp exam, usually asymptomatic but highly diagnostic. 👁️
- Optic Gliomas: Can impair vision, often detected in childhood. 👓
- Bony Abnormalities: Tibial bowing, scoliosis, pseudoarthrosis, macrocephaly, short stature. 🦴
- Hypertension: May be due to renal artery stenosis or phaeochromocytoma. 🩸
🔗 Associated Conditions
- Renal Artery Stenosis: Secondary hypertension, common in children with NF1.
- Phaeochromocytoma: Catecholamine-secreting tumour → paroxysmal hypertension, palpitations, sweats. ⚡
- CNS Tumours: Optic glioma (classic), astrocytoma, glioblastoma, ependymoma, meningioma.
- Pulmonary Fibrosis and Cardiomyopathy – rare but reported complications.
- Rare links: Medullary thyroid carcinoma in some NF1 patients, overlap with MEN syndromes is debated. 🦋
🛠️ Management
- Monitoring: Annual BP, ophthalmology review (children), dermatology/neurology surveillance for tumour burden.
- Genetic Counselling: Key for affected families (autosomal dominant inheritance, variable penetrance). 👨👩👧👦
- Multidisciplinary Care: Dermatology, neurology, ophthalmology, genetics, orthopaedics.
- Surgery: Reserved for symptomatic or function-threatening neurofibromas; plexiform lesions can be challenging to excise.
- New Therapies: MEK inhibitors (e.g. selumetinib) show promise in shrinking plexiform neurofibromas. 💊
📌 Key Exam Pearls
✅ Café-au-lait spots + axillary freckling + neurofibromas = think NF1.
✅ Always check BP in NF1 patients – hypertension may be due to renal artery stenosis or phaeochromocytoma.
✅ Lisch nodules are highly specific for NF1, best seen with slit-lamp.
✅ NF1 ↑ risk of malignant peripheral nerve sheath tumours → any rapidly enlarging/painful neurofibroma = red flag. 🚨
📖 References
Cases - Neurofibromatosis Type 1 (NF1)
- Case 1 - Child with Café-au-Lait Spots: A 7-year-old boy is referred for multiple light-brown skin patches. Exam: ≥6 café-au-lait spots >5 mm, axillary freckling, and a soft, pedunculated nodule on the arm (cutaneous neurofibroma). Ophthalmology exam reveals Lisch nodules on the iris. Diagnosis: NF1 based on NIH diagnostic criteria. Management: Multidisciplinary follow-up (neurology, dermatology, ophthalmology); annual BP monitoring (risk of renal artery stenosis); genetic counselling.
- Case 2 - Adolescent with Plexiform Neurofibroma: A 14-year-old girl with known NF1 presents with a large, soft, “bag-of-worms” mass along her neck that has been enlarging. Exam: scoliosis and multiple cutaneous neurofibromas. MRI: plexiform neurofibroma extending along the brachial plexus. Diagnosis: NF1 with plexiform neurofibroma. Management: Surgical debulking if symptomatic or compressive; regular imaging for malignant transformation risk (MPNST); psychosocial support.
Teaching Commentary 🧬
NF1 is an autosomal dominant neurocutaneous disorder (chromosome 17, neurofibromin gene). Diagnostic features (need ≥2): - ≥6 café-au-lait spots, - ≥2 neurofibromas or 1 plexiform neurofibroma, - Axillary/inguinal freckling, - Optic glioma, - ≥2 Lisch nodules (iris hamartomas), - Distinctive bone lesion (sphenoid dysplasia, tibial bowing), - First-degree relative with NF1. Complications: learning disability, hypertension (renal artery stenosis, phaeochromocytoma), scoliosis, optic gliomas, malignant peripheral nerve sheath tumours. Management: lifelong surveillance with multidisciplinary input.
Cafe au lait spots
Lisch Nodules:
| The content on this website is provided for educational and informational purposes only to support exam preparation (e.g., MLA, MRCP, USMLE) and learning. This is NOT medical advice, diagnosis, treatment, or professional guidance. It does not replace consultation with a qualified healthcare professional, official guidelines (e.g., NICE, GMC, BNF), or supervised clinical practice. Always verify information with current, authoritative sources. Makindo and its contributors accept no liability for any reliance on this content, including errors, omissions, or any resulting harm, loss, or consequences. By using this site, you agree to these terms. |
|
|
Categories
- About
- Acute Medicine
- Anaesthetics and Critical Care
- Anatomy
- Anatomy and Physiology
- Biochemistry
- Book
- Cardiology
- Collections
- CompSci
- Crib Sheets
- Critical care
- Dental
- Dermatology
- Differentials
- Drugs
- ENT
- Electrocardiogram
- Embryology
- Emergency Medicine
- Endocrinology
- Ethics
- Foundation Doctors
- GCSE
- Gastroenterology
- General Practice
- Genetics
- Geriatric Medicine
- Geriatrics
- Guidelines
- Haematology
- Hepatology
- Immunology
- Infectious Diseases
- Infographic
- Investigations
- Lists
- MRCP
- Mandatory Training
- Medical Students
- Microbiology
- Nephrology
- Neurology
- Neurosurgery
- Nutrition
- OSCE
- Obstetrics Gynaecology
- Oncology
- Ophthalmology
- Oral Medicine and Dentistry
- Orthopaedics
- Paediatrics
- Palliative
- Palliative Care
- Pathology
- Pharmacology
- Physiology
- Procedures
- Psychiatry
- Public Health
- Radiology
- Respiratory
- Resuscitation
- Revision Topics
- Rheumatology
- Statistics and Research
- Stroke
- Surgery
- Toxicology
- Trauma and Orthopaedics
- USMLE
- Urology
- Vascular Surgery