Hurler's syndrome (MPS-1)
🧬 Mucopolysaccharidoses (MPS) are a group of lysosomal storage disorders caused by deficiencies in enzymes that break down glycosaminoglycans (GAGs). This leads to progressive accumulation in tissues. Classic findings include dysostosis multiplex, coarse facies, hepatosplenomegaly, short stature, and cardiorespiratory disease.
📖 About
- Inherited metabolic disorders with progressive multi-system involvement.
- Most are autosomal recessive, except MPS II (Hunter syndrome), which is X-linked recessive.
- Incidence: rare (<1 in 100,000 births), but more frequent in certain isolated populations.
- Accumulated GAGs include dermatan sulfate, heparan sulfate, keratan sulfate, and chondroitin sulfate depending on the subtype.
🧬 Aetiology
- MPS I (Hurler / Hurler-Scheie / Scheie): Deficiency of α-L-iduronidase → accumulation of dermatan + heparan sulfate.
- MPS II (Hunter): Deficiency of iduronate sulfatase (X-linked).
- MPS III (Sanfilippo): Multiple enzyme defects affecting heparan sulfate breakdown.
- MPS IV (Morquio): Defects in enzymes breaking down keratan sulfate.
- MPS VI (Maroteaux–Lamy): Arylsulfatase B deficiency.
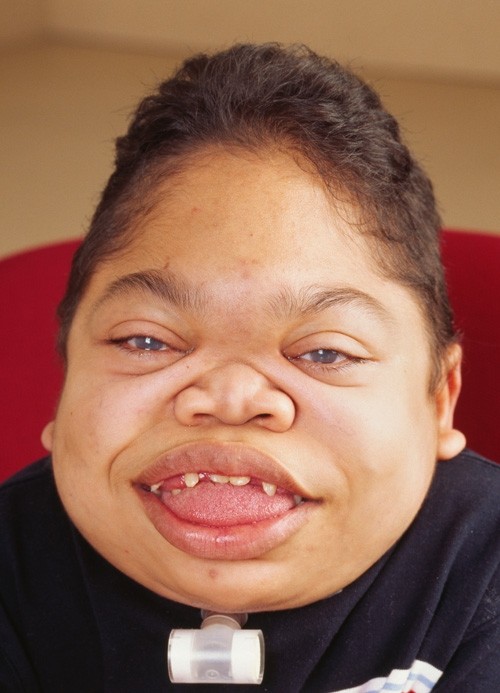
🧒 Clinical Features
- Dysostosis multiplex 🦴: Short stature, spinal gibbus, thoracic cage deformity, hip dysplasia.
- Coarse facies: Large tongue, flat nasal bridge, frontal bossing, thick lips.
- 💡 Hurler syndrome (MPS I-H): Severe developmental delay, corneal clouding, hepatosplenomegaly.
- 💡 Hunter syndrome (MPS II): Similar to Hurler but no corneal clouding; aggressive behaviour may be noted.
- Corneal clouding 👁: Seen in MPS I and VI (not in Hunter’s).
- Neurological: Developmental delay, progressive intellectual disability, hydrocephalus, behavioural issues.
- Cardiac: Valvular disease, cardiomyopathy, systemic hypertension.
- Respiratory: Upper airway obstruction, sleep apnoea, restrictive lung disease.
- Life expectancy often shortened, especially in severe phenotypes (Hurler).
🔬 Investigations
- 💧 Urine GAGs: Excess glycosaminoglycans detectable (screening test).
- 🧪 Enzyme assay: Confirmatory; measures specific lysosomal enzyme activity in leukocytes or fibroblasts.
- 🧬 Genetic testing: IDUA gene (MPS I) and others depending on subtype → definitive diagnosis and prenatal testing.
- 📷 X-rays: Dysostosis multiplex – paddle-shaped ribs, thickened calvarium, beaked vertebrae.
🩹 Management
- 💊 Enzyme Replacement Therapy (ERT): Laronidase (MPS I), idursulfase (MPS II), galsulfase (MPS VI). Improves somatic features but limited CNS penetration.
- 🧵 Haematopoietic stem cell transplant (HSCT): Considered in severe MPS I (Hurler), especially before 2 years, to slow neurodegeneration.
- ⚕️ Supportive care: ENT (adeno-tonsillectomy, tracheostomy), orthopaedics (joint replacement, spinal surgery), ophthalmology (corneal transplant), cardiology follow-up.
- 🧘 Physio & OT: Improve mobility, respiratory physiotherapy for secretion clearance.
- ❤️ Palliative support in advanced disease stages.
📊 Exam Pearls
- 👁 Corneal clouding = Hurler, Scheie, Maroteaux–Lamy. ❌ Absent in Hunter.
- 👦 Hunter syndrome = “Hunter has no clouded eyes.” (X-linked, behavioural issues).
- 📷 Radiology buzzword: Dysostosis multiplex.
- 🧬 Screening siblings is crucial due to autosomal recessive inheritance.
📚 References
