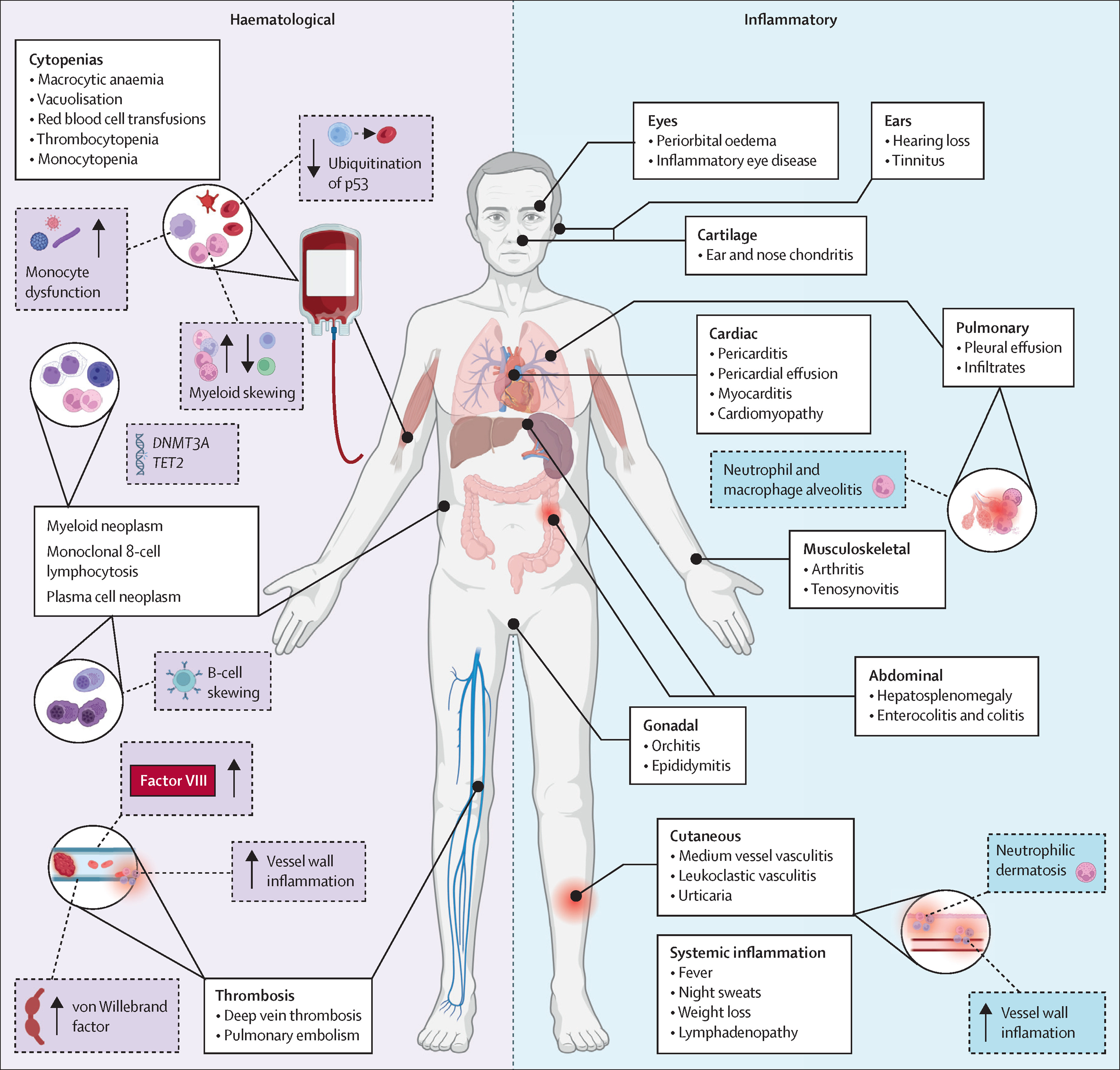
VEXAS syndrome (Vacuoles, E1 enzyme, X-linked, Autoinflammatory, Somatic) is a late-onset, treatment-refractory autoinflammatory disorder caused by somatic (acquired) mutations in UBA1 within haematopoietic stem/progenitor cells, producing a combined systemic inflammatory + bone-marrow (haematological) phenotype.
🔑 Key points
- Think VEXAS in an older man with recurrent fevers/inflammation, very high CRP, and macrocytic anaemia (often ± thrombocytopenia/leukopenia), especially if “vasculitis/chondritis/Sweet syndrome” keeps relapsing when steroids are reduced.
- It is genotype-defined (UBA1 mutation). Clinical labels may shift over time (e.g., relapsing polychondritis phenotype → vasculitis phenotype → MDS phenotype).
- Bone marrow often shows cytoplasmic vacuoles in myeloid/erythroid precursors (supportive, not diagnostic on its own).
🧬 Pathophysiology
UBA1 encodes the major E1 ubiquitin-activating enzyme that initiates ubiquitination, a key pathway for protein quality control and immune signalling. Somatic UBA1 mutations create an abnormal
myeloid inflammatory clone, driving cytokine-heavy innate immune activation (systemic inflammation) while also impairing haematopoiesis (macrocytosis, cytopenias, MDS-like features). This dual biology
explains why patients can look rheumatological and haematological at the same time, and why isolated immunosuppression often struggles to achieve durable control.
👀 Clinical features
| Domain |
Typical manifestations |
Clinical pearls |
| Constitutional |
Recurrent fevers, weight loss, severe fatigue; markedly raised CRP/ESR |
Often “sepsis-like” inflammation with negative cultures; relapses when pred is tapered |
| Skin |
Neutrophilic dermatoses (Sweet-like), vasculitic rash, nodules |
Biopsy may show neutrophilic inflammation and/or vasculitis; helps exclude infection/drug reactions |
| Cartilage |
Auricular/nasal chondritis, hoarseness/stridor if laryngo-tracheal involvement |
“Relapsing polychondritis” phenotype in an older man + macrocytosis is a big clue |
| Pulmonary/serosal |
Inflammatory infiltrates, pleuritis/effusions; dyspnoea |
Repeated “pneumonia” diagnoses with high inflammatory markers should trigger reconsideration |
| Vascular/thrombotic |
Vasculitis (variable vessel size), thrombosis (VTE) |
Thrombosis risk can be substantial; assess and manage VTE risk proactively |
| Haematology |
Macrocytic anaemia; cytopenias; MDS-like changes |
Macrocytosis out of proportion to B12/folate issues is common; consider marrow + genetics early |
🧪 Investigations
- Baseline labs: FBC (look for macrocytosis/cytopenias), film, reticulocytes; CRP/ESR; U&E/LFTs; ferritin; LDH; B12/folate/TSH; haematinics.
- Autoimmune/vasculitis screen (as guided by phenotype): ANA/ENA, complements, ANCA, RF/CCP, cryoglobulins; hepatitis B/C and HIV where relevant.
- Infection exclusion: cultures, viral PCRs, imaging as indicated (VEXAS is often mis-treated as infection).
- Imaging: CT chest/abdomen/pelvis or PET-CT may help map inflammatory foci and exclude malignancy/infection (local protocol-driven).
- Bone marrow: aspirate/trephine if cytopenias/macrocytosis or suspicion of MDS; look for vacuoles in precursors and dysplasia; send cytogenetics/NGS as per haem pathway.
- Definitive test: UBA1 mutation testing on peripheral blood or marrow (liaise with Haematology/Clinical Genetics/local molecular lab route).
✅ Diagnostic approach (practical UK workflow)
- Recognise the pattern: refractory systemic inflammation + macrocytic anaemia/cytopenias ± chondritis/skin/pulmonary/vasculitic features.
- Exclude mimics urgently: infection (including endocarditis/TB), malignancy, classic systemic vasculitides, adult-onset Still’s, drug reactions.
- Engage Rheumatology + Haematology early: this is a cross-specialty disease; avoid prolonged steroid monotherapy.
- Confirm with genetics: send UBA1 testing; marrow findings support but do not replace genetic confirmation.
🔁 Differentials to actively consider
- Relapsing polychondritis (non-VEXAS)
- ANCA-associated vasculitis / PAN / other systemic vasculitides
- Adult-onset Still’s disease
- Sweet syndrome / neutrophilic dermatoses (secondary causes)
- Myelodysplastic syndrome without systemic autoinflammation
- Occult infection (endocarditis, TB, deep-seated abscess)
- Haematological malignancy with inflammatory paraneoplastic features
💊 Management (principles)
There is no single “one-size-fits-all” regimen. Management balances rapid inflammation control (often steroid-responsive initially) with steroid-sparing therapy and, where appropriate,
treatments aimed at the haematopoietic clone. Patients should be managed in a joint Rheumatology–Haematology model, with early consideration of complications (infection, thrombosis,
cytopenias, organ involvement).
| Treatment layer |
What’s used |
Notes / cautions |
| Induction / flare control |
Glucocorticoids (often effective short-term) |
Relapse on taper is common; avoid long-term high-dose dependence; assess bone protection, glucose, BP |
| Steroid-sparing anti-inflammatory |
Biologics or targeted agents (case-series experience: IL-6 pathway inhibitors, JAK inhibitors, IL-1 blockade in selected cases) |
Evidence base evolving; infection risk is significant-vaccination, screening, and prophylaxis as per local policy |
| Clone / marrow-directed |
Haematology-led therapies for associated MDS (supportive care, transfusions, EPO where appropriate, disease-modifying options per MDS subtype) |
Optimise haematinics; monitor iron overload if transfusion-dependent |
| Potentially curative |
Allogeneic haematopoietic stem cell transplantation (HSCT) in carefully selected patients |
High-risk pathway; requires specialist MDT assessment (fitness, donor, disease trajectory, comorbidity) |
| Supportive / prevention |
Thrombosis risk management; bone protection; infection prevention; GI protection if indicated |
Low threshold for VTE assessment; consider PJP prophylaxis if on prolonged high-dose steroids/combined immunosuppression (local guideline) |
🩺 Monitoring & follow-up
- Inflammation: symptoms, temperature, CRP/ESR trends (watch for discordance if infection emerges).
- Haematology: serial FBC, MCV, transfusion needs; marrow reassessment if cytopenias evolve.
- Complications: VTE surveillance, infection vigilance, steroid toxicity (bone, metabolic, psychiatric), cardiovascular risk.
- Organ involvement: pulmonary function/imaging, ENT review if airway chondritis suspected, dermatology input for biopsy/phenotyping.
🚨 Red flags needing urgent escalation
- Stridor/voice change or airway symptoms (possible laryngo-tracheal chondritis)
- Rapidly progressive cytopenias or bleeding
- Severe hypoxia / new pulmonary infiltrates (infection vs inflammation)
- Suspected thrombosis (PE/DVT, atypical sites)
- Persistent high fever with immunosuppression (treat as infection until proven otherwise)
📚 References
- Beck DB, et al. Somatic Mutations in UBA1 and Severe Adult-Onset Autoinflammatory Disease. New England Journal of Medicine (2020). (VEXAS original description).
- NEJM Editorial: VEXAS Syndrome (context and interpretation of discovery).
- Rheumatology review (OUP): recent clinical updates and management considerations.